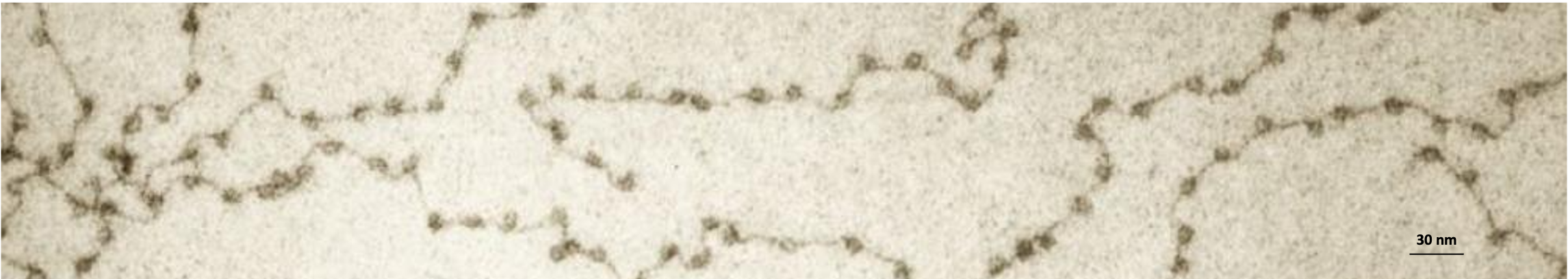
Adapted from American Scientist
Donald E. Olins, Ada L. Olins. 1978
How do genomes encode dynamic gene activity?

Since the discovery of the lac operon in the 1960s it became apparent that DNA sequences can harbor regulatory instructions, guiding transcription of a target gene. Though a seemingly static entity, the DNA has the capacity to instruct highly dynamic and cell type-specific gene activity patterns, that underlie complex biological processes such as developmental progression in multicellular organisms.
Key agents in this process are transcription factors (TFs), whose timely binding to regulatory sequences induces activation or repression of a target gene. Yet, even in the relativity simple yeast genome, the abundance and sequence preferences of TFs are insufficient to fully account for changes in gene activity.
What are the other determinants within regulatory sequences that allow modulation of transcription?
Studies in recent years made it abundantly clear that in multicellular organisms a gene can be regulated by multiple DNA sequences. Such sequences, namely enhancers, can reside at varying genomic distances from the target gene, and a single enhancer can activate multiple genes. Forging the right regulatory interactions in this genomic maze is necessary to ensure that the right genes are expressed at the right level, in specific cell types, and at specific times.
We strive to understand how regulatory information is distributed amongst multiple DNA sequences, what governs the specificity of regulatory interactions and how a gene integrate regulatory instructions form multiple sources, or across large genomics distances.
The inner workings of individual regulatory sequences
By employing massively parallel reporter assays (MPRA), in previous work, we were able to identify determinants of gene activity within regulatory sequences, going beyond the classically studied TF core motifs. We developed methods to allow measurements of gene activity and protein binding across thousands of systematically designed regulatory sequences. These measurements showed how sequences flanking core motifs can alter TF binding. Machine learning models predicted this differential binding based on the flanking sequences composition or, interestingly, based on features of DNA shape.
To uncover co-occuring binding events on regulatory sequences, we devised a methodology that couples MPRA with DNA-recorded single-cell footprinting (via exogenous methylation). This provided means to quantify differential chromatin sensitivity/remodeling across TFs.
Models, informed by these measurements, help elucidate how features of regulatory sequences (composition and motif ‘grammar’) dictate TF-nucleosome interplay, collaborative TF control and consequently gene activity.
Our lab is working on further developing these approaches, now going beyond the investigation of individual regulatory sequences. We are also continuing to explore the interplay between the local chromatin environment and 3D chromatin conformations (see below) to transcription.

Disentangling long-range regulatory interactions
We now know that eukaryotic genomes are replete with long-range regulatory interactions. These are thought to rely on physical DNA interactions within individual cells. Yet both the spatial scale and the time scale of these physical interactions remains unclear.
Are these spatiotemporal parameters regulated across cell types and developmental times? Are these rate-limiting for regulatory functional interactions? What governs the specificity of physical and/or functional interactions, and what are the molecular mechanisms that facilitate information transfer from these genomically disparate DNA elements?
Our lab employs both genomics and imaging approaches to address these questions. For example, with the aid of CRISPR-based genome editing we monitor real-time endogenous long-range physical interactions and the functional readout, namely transcription. We then perturb the potentially underlying regulatory determinants.



On the bursty road from transcription regulation to transcriptional output
Transcription has been shown to occur in bursts in various systems, from bacteria to mammalian cells. The amount of mRNA produced over any time interval depends on successive transcription initiation events (within ‘ON’ periods), interspersed by quiescent (‘OFF’) periods. Yet the source of these bursts remains unclear. Nor do we understand how their kinetic properties are modulated by regulatory processes to quantitatively tune mRNA production.
We explore quantitive properties of transcriptional bursting and what these can tell us about the molecular mechanisms underlying transcription initiation and the regulatory processes modulating these molecular events.

Our lab uses a combination of experimental and computational approaches to study
gene regulation
across spatiotemporal scales